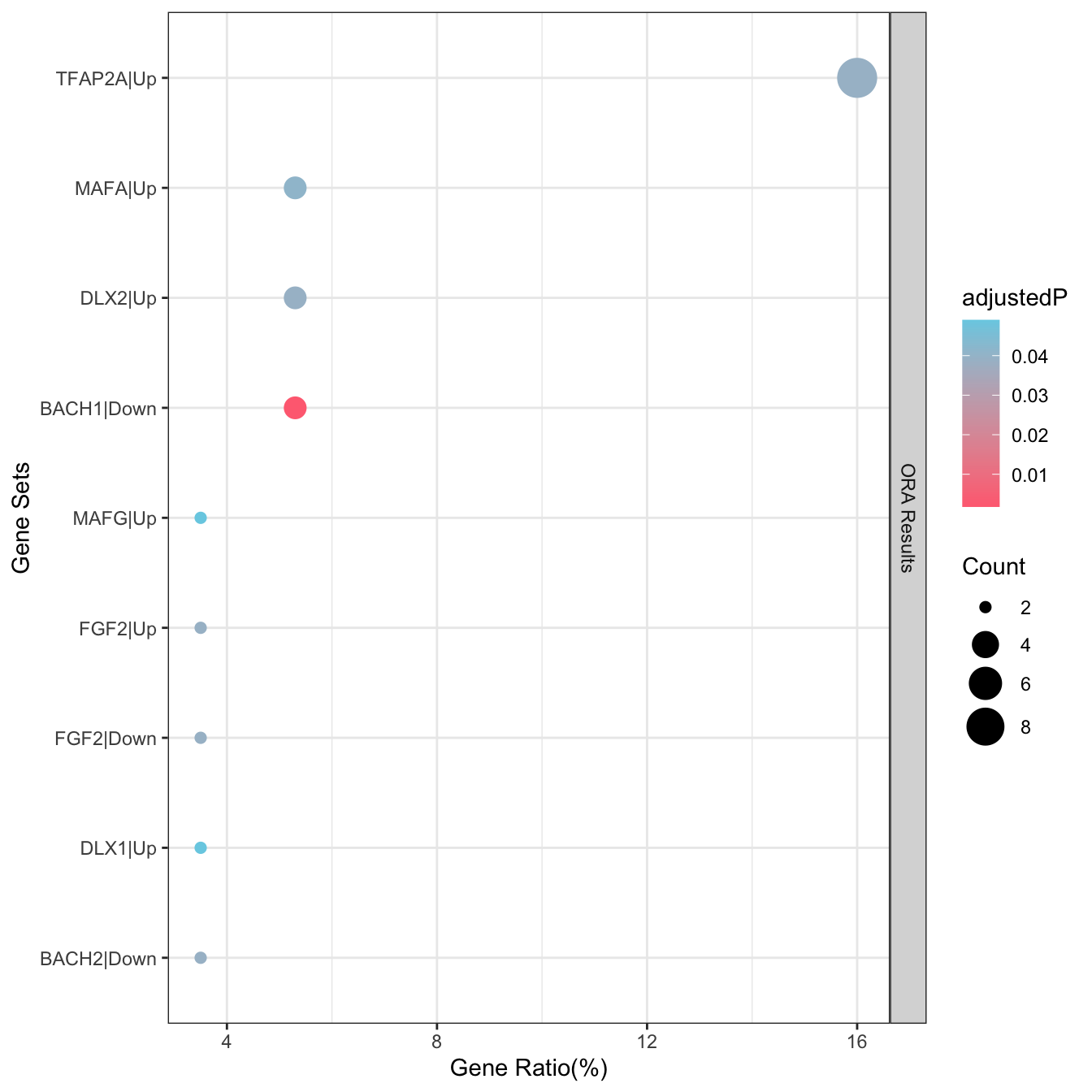
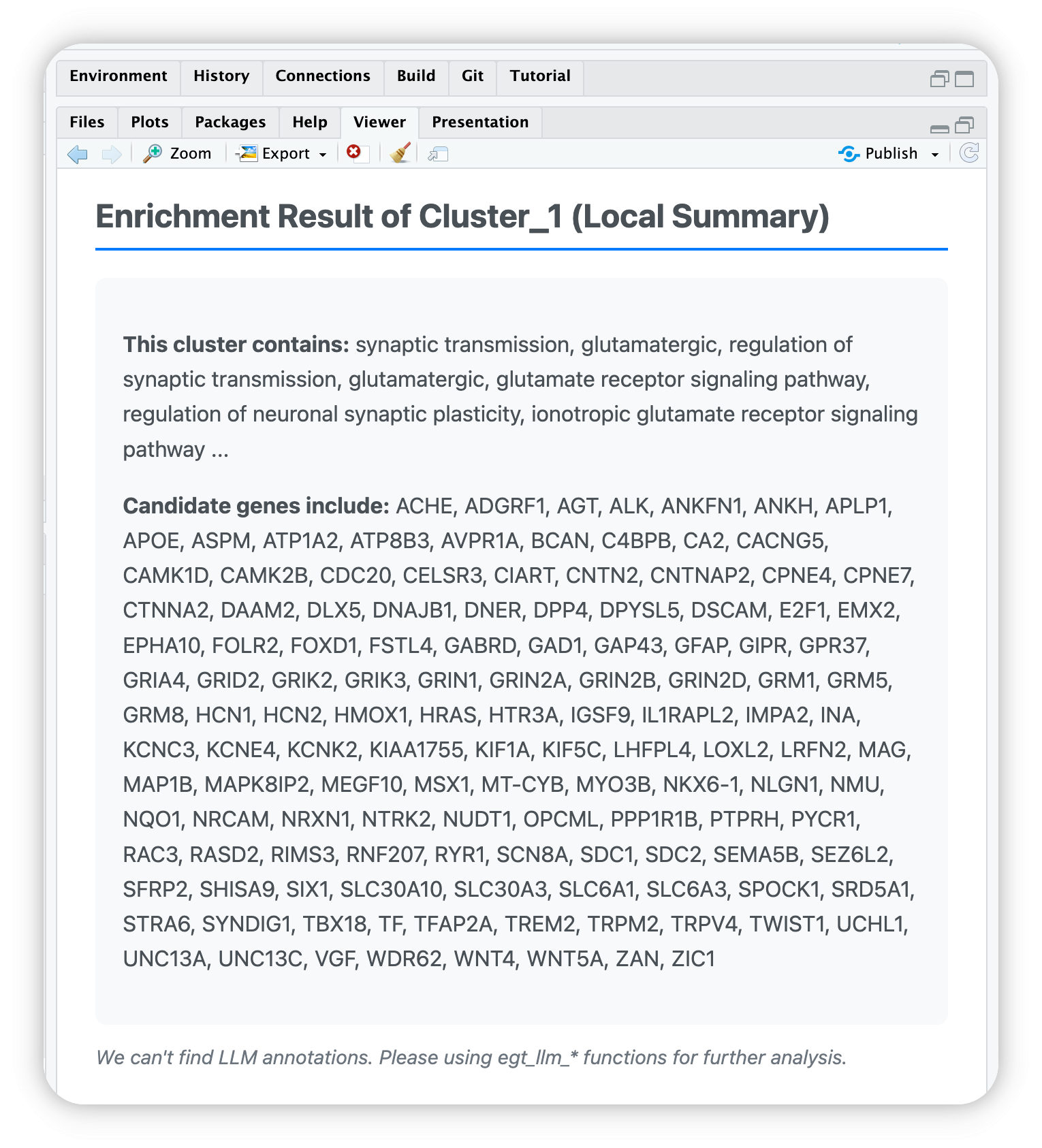
• Candidate genes includes ACHE, ADGRF1, AGT, ALK, ANKFN1, ANKH, APLP1, APOE,
ASPM, ATP1A2, ATP8B3, AVPR1A, BCAN, C4BPB, CA2, CACNG5, CAMK1D, CAMK2B, CDC20,
CELSR3, CIART, CNTN2, CNTNAP2, CPNE4, CPNE7, CTNNA2, DAAM2, DLX5, DNAJB1, DNER,
DPP4, DPYSL5, DSCAM, E2F1, EMX2, EPHA10, FOLR2, FOXD1, FSTL4, GABRD, GAD1,
GAP43, GFAP, GIPR, GPR37, GRIA4, GRID2, GRIK2, GRIK3, GRIN1, GRIN2A, GRIN2B,
GRIN2D, GRM1, GRM5, GRM8, HCN1, HCN2, HMOX1, HRAS, HTR3A, IGSF9, IL1RAPL2,
IMPA2, INA, KCNC3, KCNE4, KCNK2, KIAA1755, KIF1A, KIF5C, LHFPL4, LOXL2, LRFN2,
MAG, MAP1B, MAPK8IP2, MEGF10, MSX1, MT-CYB, MYO3B, NKX6-1, NLGN1, NMU, NQO1,
NRCAM, NRXN1, NTRK2, NUDT1, OPCML, PPP1R1B, PTPRH, PYCR1, RAC3, RASD2, RIMS3,
RNF207, RYR1, SCN8A, SDC1, SDC2, SEMA5B, SEZ6L2, SFRP2, SHISA9, SIX1, SLC30A10,
SLC30A3, SLC6A1, SLC6A3, SPOCK1, SRD5A1, STRA6, SYNDIG1, TBX18, TF, TFAP2A,
TREM2, TRPM2, TRPV4, TWIST1, UCHL1, UNC13A, UNC13C, VGF, WDR62, WNT4, WNT5A,
ZAN, ZIC1 (We will print all genes. Please stroll to top to read)